Tratamiento del dolor crónico intenso
Dra. Esperanza Ortigosa - Hospital Universitario de Getafe
Dra. Estefanía Limeres - Hospital Universitario Lucus Augusti de Lugo
Dr. Jesús Estrada - Hospital Universitario de Getafe
INTRODUCCIÓN
El dolor a pesar de ser tan antiguo como el hombre, no es fácil de definir. En 1979 la IASP (Asociación Internacional para el Estudio del Dolor) lo define como “una experiencia sensorial y emocional desagradable, asociada con una lesión hística, presente o potencial, o descrita en términos de la misma”[1]
En esta definición se puede entrever que el dolor no es un mero síntoma sensorial, sino que debe entenderse como una experiencia aversiva perceptual y afectiva compleja, determinada tanto por las respuestas biológicas a los estímulos nociceptivos como por el significado de esos estímulos para cada sujeto, con interrelación de múltiples factores, tanto físico-sensoriales como psicológicos, emocionales y subjetivos.
Precisamente en esta variedad de factores que influyen sobre la vivencia del dolor por cada paciente y en el hecho que cada paciente experimenta el dolor de una forma propia, radica la necesidad de un enfoque terapéutico personalizado[2]).
CLASIFICACIÓN DEL DOLOR
Los criterios de clasificación del dolor son múltiples. A continuación se sistematizan los tipos de dolor en función de varios de dichos criterios[3]:
- Según las estructuras que pueden dar lugar al origen del dolor: somático y visceral.
- Según los mecanismos neurofisiológicos: nociceptivo y neuropático.
- Según la duración: agudo y crónico.
Dolor somático
Dolor originado en la piel, músculos, articulaciones, ligamentos o huesos. Se caracteriza por ser selectivo, metamérico y no referido. Hay participación de nociceptores específicos y del sistema nervioso periférico.
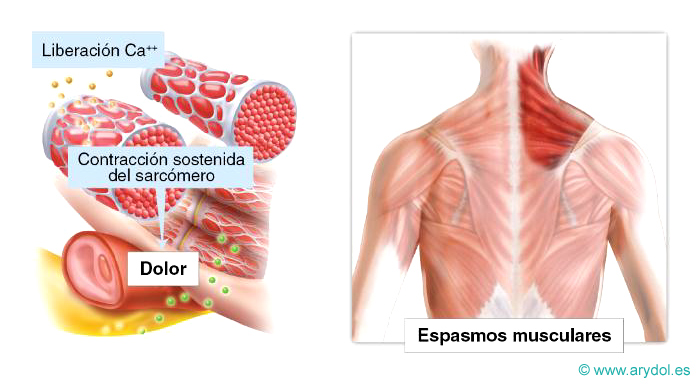
Fig. 1
Dolor visceral
Dolor originado en los órganos internos. Es la forma de dolor que, con mayor frecuencia, aparece como consecuencia de enfermedades. Desde el punto de vista fisiopatológico los estímulos principales que lo desencadenan son: distensión o dilatación brusca, espasmo o contracción del músculo liso en particular, si hay isquemia, e Irritantes químicos.
Hay participación de nociceptores inespecíficos y del sistema nervioso autónomo.
Clínicamente, se caracteriza por: ser sordo y mal localizado. A menudo, se refiere a la superficie del organismo en zonas distantes de la víscera que lo origina, siguiendo las leyes de la organización segmentaria.
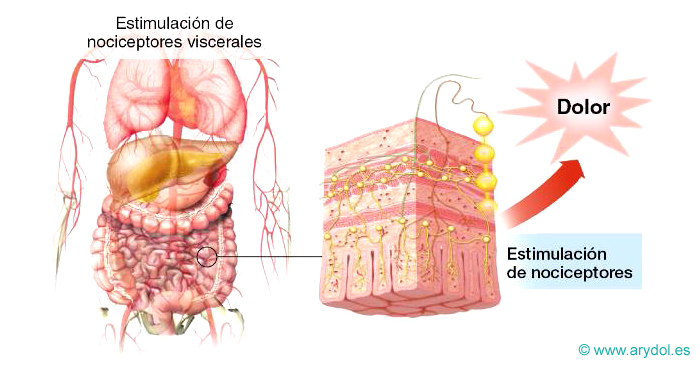
Fig. 2
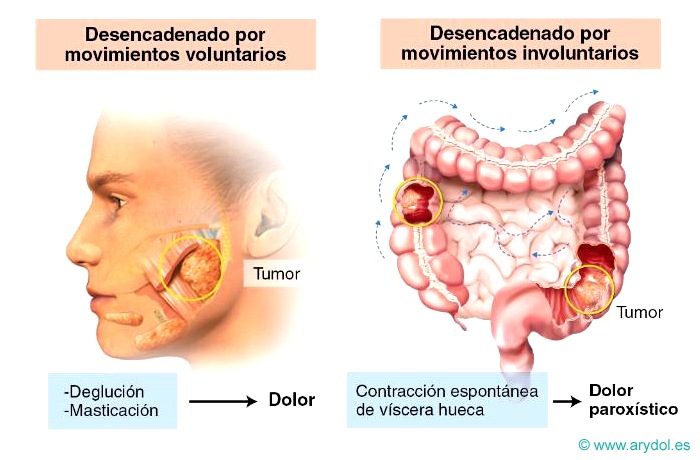
Fig. 3
Dolor nociceptivo
Es el dolor que aparece como consecuencia de la aplicación de estímulos que producen daño o lesión en órganos somáticos o viscerales. Frecuentemente, cumple una misión de protección.
Es aquel dolor que aparece en los individuos normales después de un estímulo que produce daño o lesión. Se denomina también dolor normal o sensorial, y es el resultado último de la activación de un sistema sensorial específico que abarca nociceptores específicos, vías ascendentes y córtex cerebral. Ej. El dolor percibido ante un pinchazo con una aguja.
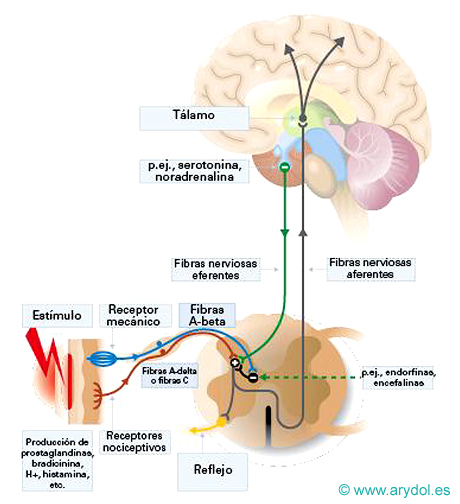
Fig. 4
Esquema simplificado del dolor nociceptivo
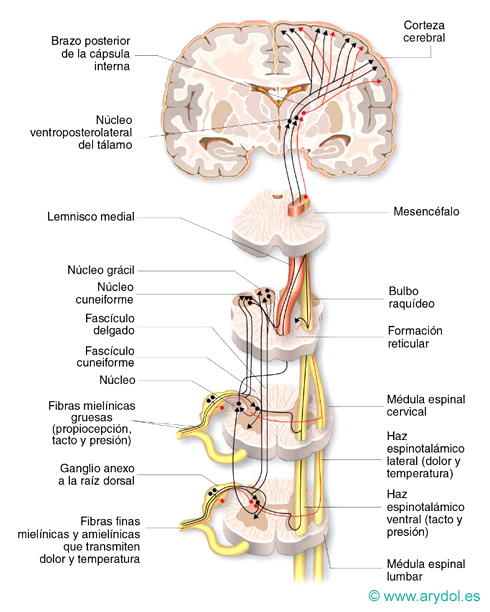
Fig. 5
La vía del dolor
Dolor neuropático
Se denomina también dolor anormal o patológico[4]. Sólo aparece en una minoría de los individuos (aunque su prevalencia es mayor de lo que se pensaba) y está motivado por enfermedad o lesión del sistema nervioso central o periférico. Es un dolor persistente, rebelde al tratamiento, a veces de aparición tardía después de la lesión. Se observa una reacción anormal del sistema nociceptivo hasta tal punto que a veces, hay ausencia total de relación causal entre la lesión tisular y el dolor5. La cascada desencadenante implica una sensibilización de los receptores periféricos y/o centrales, de manera que envían al córtex sensaciones de dolor ante estímulos que habitualmente no son dolorosos. Los receptores quedan así en estado de “hipersensibilización”, emitiendo descargas que serían las responsables del dolor lancinante y paroxístico, transmitido por fibras A-beta. Un buen ejemplo, es el dolor por avulsiones traumáticas de plexos nerviosos.
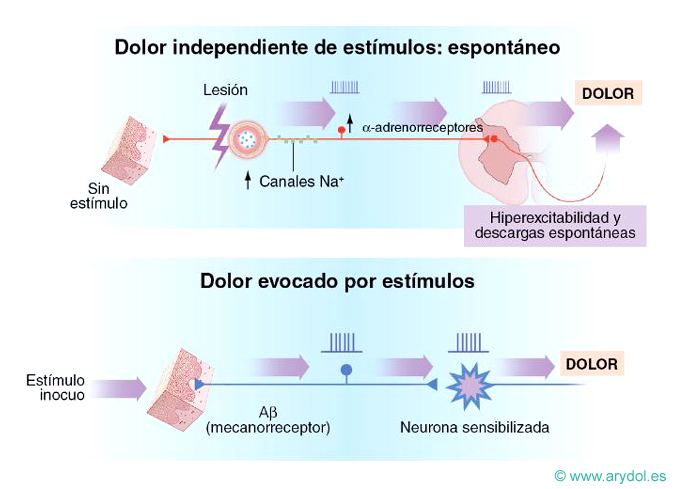
Fig. 6
Dolor neuropático
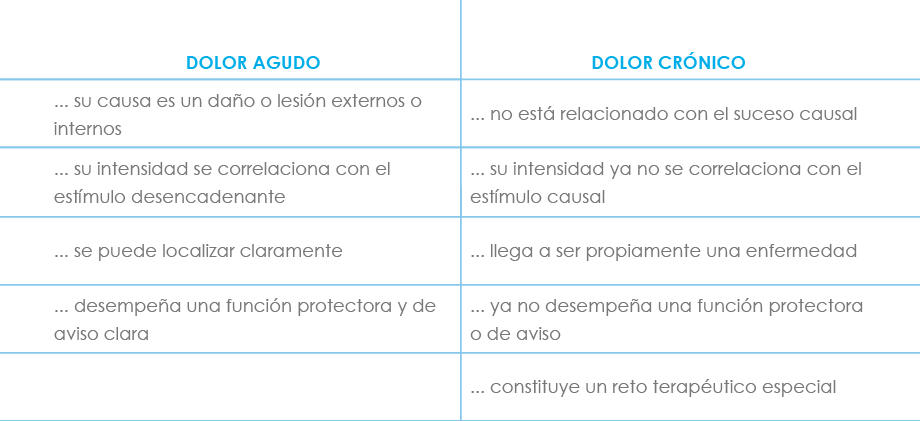
Dolor agudo
Es aquel dolor que no suele durar más de lo que tarda en resolverse la lesión causante y en todo caso, menos de un período, arbitrariamente establecido, de entre 3 y 6 meses.
Dolor crónico
El dolor crónico se define como aquel que persiste más allá de 6 meses (IASP) a pesar de la desaparición de las causas que lo originaron. Bonica lo define como un dolor que persiste al menos un mes más que la lesión causal y que permanece una vez que dicha lesión desaparece.
Llegado a este punto, el dolor crónico pierde el carácter de aviso o alarma protectora del dolor agudo y se torna en un problema en sí mismo, convirtiéndose en una enfermedad propia o en parte importante de ésta. Es frecuente la aparición de alteraciones y trastornos emocionales (labilidad, depresión), conductuales, del sueño y del apetito, deterioro físico progresivo, respuesta neuroendocrina al estrés atenuada (o ausente), disminución de la actividad y con frecuencia efectos secundarios de los tratamientos administrados.
Al igual que el dolor agudo, suele producirse por una lesión, pero se perpetúa por efectos patógenos o físicos independientemente de la causa original. Afecta a casi un 20% de la población en nuestro país y de ellos, un tercio padece un dolor intenso. Es un síndrome de difícil control y muy frecuente en diversas patologías: artritis, fracaso de la cirugía vertebral, lesiones y accidentes, patología oncológica, SIDA y trastornos del sistema nervioso… Todo ello conduce a menudo a que el dolor se convierte en el aspecto esencial de la vida del paciente, cuya existencia gira en torno a éste, en ocasiones de forma dramática.
DIAGNÓSTICO Y EVALUACIÓN
El manejo adecuado del dolor depende de un diagnóstico y evaluación adecuado. Habrá que tener en cuenta el historial médico general del paciente, el historial del dolor y el historial psicológico.
Así mismo como en cualquier otra patología se realizarán las pruebas analíticas electrofisiológicas y de imagen que sean necesarias para establecer un diagnóstico y un tratamiento personalizado.
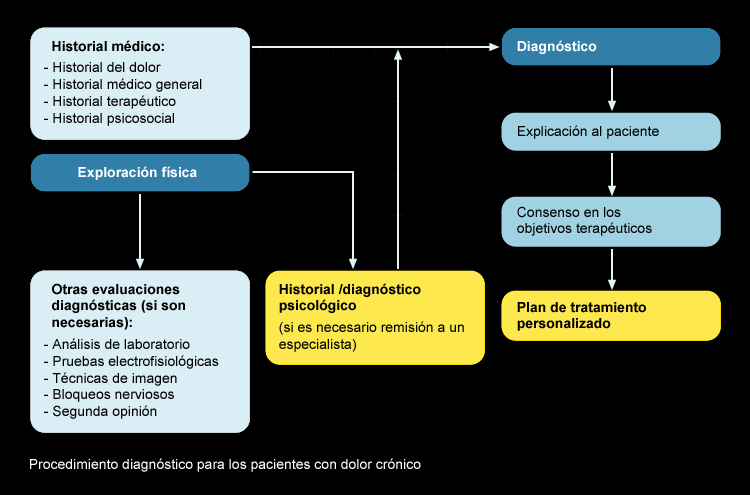
Una de las dificultades mayores que nos encontramos es la aplicación de un medio para valorar y cuantificar el dolor.
En este sentido tenemos varias escalas unidimensionales que valoran la intensidad del dolor con independencia de las demás características en ese sentido podemos citar:
1) Escala de intensidad numérica[6].
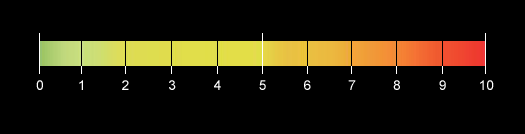
Sin embargo este tipo de escala no es útil para todos los pacientes[7,8,9]. Los estudios muestran que en pacientes ancianos las escalas basadas en descriptores verbales han resultado más útiles que las visuales (Visual Analogic Scale/Escala Visual Analógica). Las razones de estas diferencias no están claras, sin embargo podría explicarse con la dificultad de comprensión que a veces afecta a este grupo etario.
2) Escala de intensidad descriptiva.
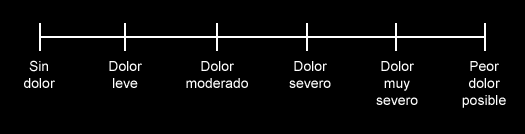
3) Escala de valoración de caras de dolor de Wong-Baker.

Escalas multidimensionales
Las escalas multidimensionales incorporan conceptos que permiten una amplia caracterización del dolor en un único paso y valoran su repercusión en la esfera socio-laboral y familiar del paciente. Ejemplo de escala multidimensional de valoración del dolor: agenda del dolor[9].
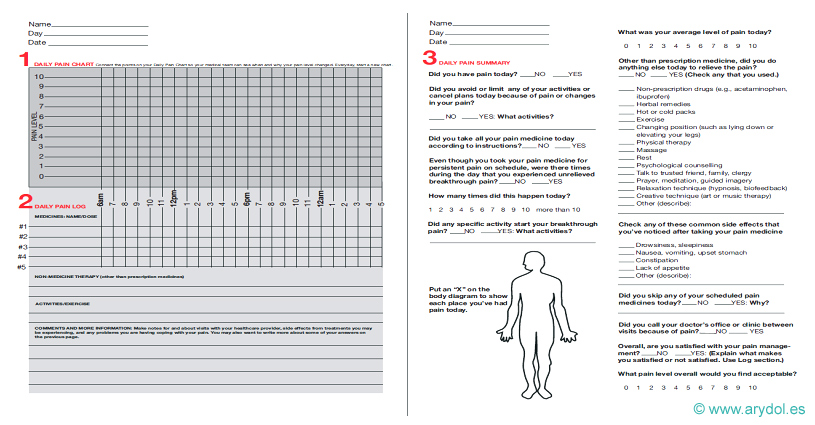
Fig. 12
TRATAMIENTO
Los analgésicos opiáceos constituyen los fármacos de elección para el dolor crónico moderado o intenso. En casos más rebeldes recurrimos a técnicas invasivas. Cuando hablamos de dolor crónico intenso debemos recordar el concepto “ascensor” de la OMS, sustituyéndolo por el habitual “escalera analgésica”; y es que si pretendemos atajar un dolor con una EVA >6, debemos alcanzar los niveles más altos de inicio; siendo en estos casos los opioides débiles insuficientes incluso a dosis máximas.
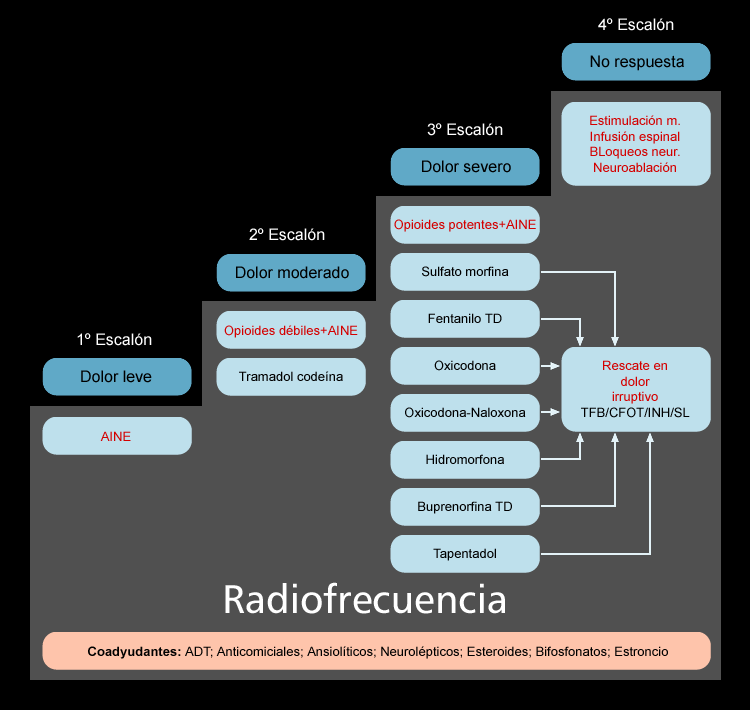
Los principales efectos secundarios son leves (somnolencia, mareos, náuseas) y transitorios, y no precisan tratamiento; no así el estreñimiento que siempre debe tratarse dada su inevitable aparición, en especial en pacientes encamados o aquellos con tumores del aparato digestivo. Otros efectos secundarios como depresión respiratoria, alucinaciones o retención urinaria son extremadamente raros, pero nos obligan a replantear el tratamiento. Su uso estará contraindicado si existe la posibilidad de otro tratamiento eficaz, la respuesta a los opioides es escasa o nula, la escalada de dosis es incontrolable, aparecen efectos adversos graves con dosis bajas, el paciente padece patología psiquiátrica severa o es imposible realizar un seguimiento estrecho.
Si realizamos una correcta selección del paciente, la adicción aparece en un bajo porcentaje (0.5 – 1%), debiendo iniciar la terapia en consenso con el paciente y la familia; Antes de iniciar el tratamiento debemos pues evaluar la personalidad del paciente, asegurar naturaleza orgánica de dolor, descartar patología psiquiátrica grave, deterioro socio-laboral importante, acordar de mutuo acuerdo con el paciente el tratamiento (escrito), comprobar la ausencia por parte de éste de adicción a drogas y por último evaluar su sensibilidad a opioides.

Comenzaremos a dosis bajas, aumentando un 50% cada 48 horas de forma progresiva, al igual que su retirada.
El inicio con opioides mayores correspondería al tercer escalón donde están fármacos como fentanilo (transdérmico, transmucoso, inhalado, sublingual), buprenorfina (transdérmico), metadona, oxicodona, hidromorfona, tapentadol y morfina, que describiremos a continuación.
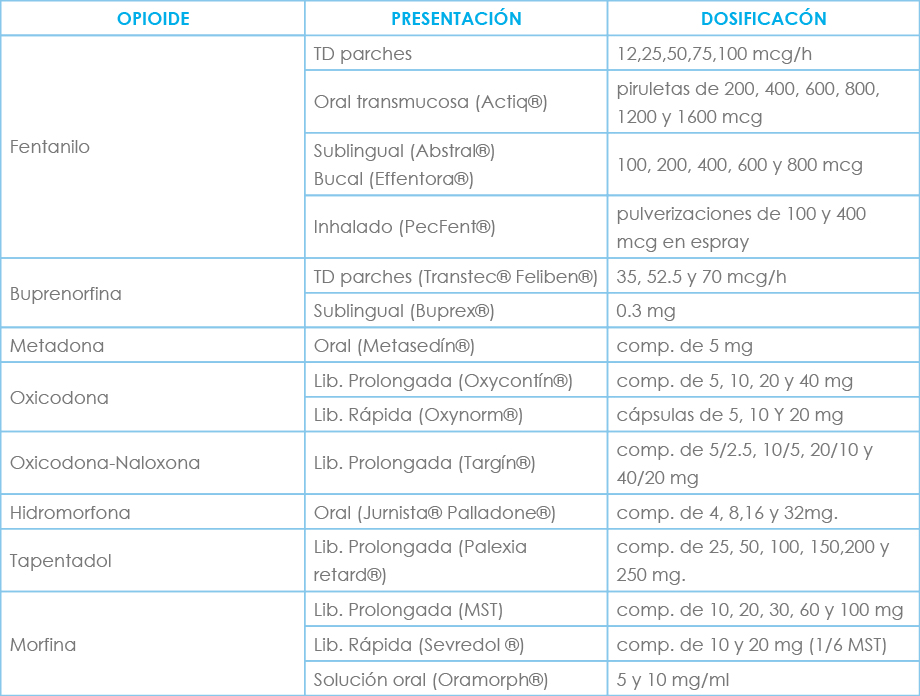
Morfina
La morfina es el prototipo de opiáceo con el que se comparan los demás. Su principal acción es la analgésica. Su unión a receptores µ, situados en el sistema nervioso y en la periferia y la amplia distribución de los mismos es responsable de sus diferentes acciones farmacológicas[10,11,12]. La morfina es absorbida rápidamente tras su administración por cualquier vía oral, iv, im…, alcanzando picos plasmáticos por vía IV o IM a los 20-30 minutos. Su vida media de eliminación oscila entre 1,7-3,3 horas. Sufre metabolización hepática obteniéndose dos metabolitos principales: la morfina-3-glucurónido (M3G) y la morfina-6-glucurónido (M6G), y en menor proporción normorfina13. Un 10% se elimina por vía renal sin metabolizar. La M6G tiene una elevada afinidad por el receptor µ, su potente acción analgésica contribuye al efecto analgésico final de la morfina. La M3G se ha asociado a fenómenos de tolerancia, hiperalgesia y alodinia tras tratamiento con dosis elevadas de morfina[14,15].
La dosis IV varía entre 0,01-0,2 mg/kg. Por vía IM, su farmacocinética es más predecible cuando se administra en la musculatura deltoidea. Es un fármaco hidrofílico y, por tanto, de acción más retardada que los lipofílicos (fentanilo). Por vía espinal alcanza fácilmente la estructuras encefálicas por lo que ha de tenerse muy en cuenta la posibilidad de provocar depresión respiratoria tardía, extremando la vigilancia. Como todos los opioides, en mayor o menor grado, puede liberar histamina y provocar prurito, situación que puede revertirse con dosis bajas de naloxona.
Fentanilo
Opioide lipofílico, lo que le permite un rápido paso a través de la membrana hematoencefálica y una mayor rapidez de acción. 100-150 veces más potente que morfina. Menor posibilidad de ascensión rostral cuando se utiliza por vía espinal y menor incidencia de depresión respiratoria tardía.
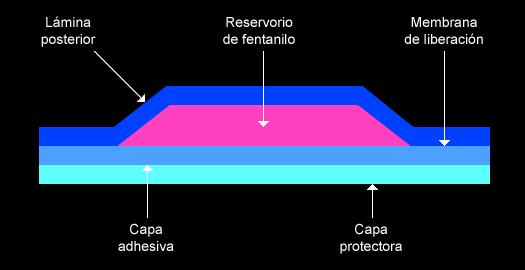
El fentanilo fue el primer opioide disponible en vía transdérmica para su administración crónica y por esta vía pierde la ventaja de su rapidez de acción (su efecto pico aparece unas 12 horas después de su colocación) circunstancia que debe advertirse siempre al paciente.
El parche reservorio está formado por cuatro láminas superpuestas sobre una capa protectora.
- Lámina posterior (poliéster): barrera para evitar pérdidas de sustancia activa de fentanilo.
- Reservorio: contiene fentanilo en forma de gel, proporciona una liberación continua de fentanilo durante 72 horas (este opioide es el único componente farmacológicamente activo).
- Membrana: regula la liberación de fentanilo a la superficie cutánea.
- Capa protectora: recubre la parte adhesiva del parche mientras este permanece dentro de la bolsa, se retira antes de aplicar el parche.
La absorción del fármaco por esta vía puede aumentar hasta en un 30% en diversas situaciones como la aplicación de fuentes de calor externas (por ejemplo una manta eléctrica), fiebre, la temperatura de la piel o su estado (rasguños), la zona del cuerpo o la grasa corporal; lo que ocasiona variabilidad en la duración del efecto e incrementa la toxicidad[16].
El reservorio además tiene varias desventajas: una importante variación en las dosis liberadas, fugas de fentanilo por el daño en el reservorio, una adhesión a la piel que no es ideal y uso ilícito por la facilidad de extraer el fármaco del reservorio y su alto contenido. Por ello, han surgido nuevos tipos de parches transdérmicos denominados matriciales que modifican el sistema controlador de liberación; el principio activo se encuentra incluido en una matriz, a través de la cual difunde hacia la piel sin que exista una membrana de control. En Durogesic matrix y Fentanilo TD el fármaco está disuelto en una formulación semisólida de un adhesivo de metacrilato; esto complica la extracción con fines ilícitos y anula el riesgo de fuga accidental; Las dosis de fentanilo administradas pueden ser incluso superiores a las del reservorio, puede ser cortado (aunque esto es controvertido) y es transparente.
Más tarde surgió una segunda generación (Fendivia, Matrifén) que incluye una membrana de control de la velocidad de liberación situada entre la matriz y el revestimiento y capa adhesiva; así se mejora el control de liberación, hay mayor estabilidad en la concentración de fentanilo durante las 72 horas y permite que la liberación de FTD sobre la superficie cutánea sea más lenta que su difusión a través de la microcirculación[17], sólo un 30 % del fentanilo total se libera en las primeras 4 horas en estos parches frente al 90 % en el resto.
Los parches matriciales de 2ª generación son bioequivalentes con los del sistema reservorio.
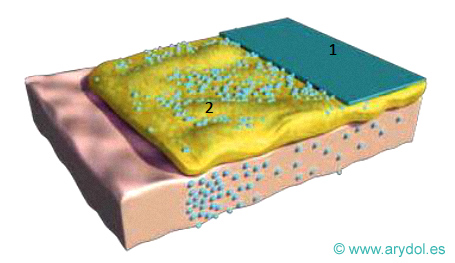
Fig. 15
Parche matricial sin membrana de control de la velocidad de liberación
1 Lámina de soporte exterior
2 Matriz con adhesivo y fentanilo
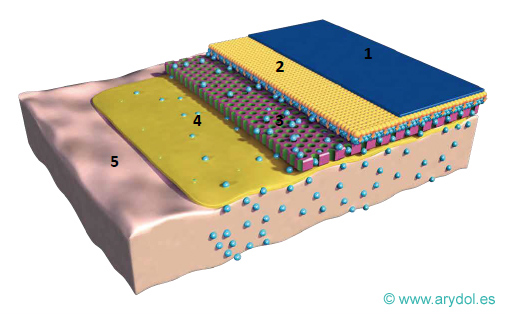
Fig. 16
Parche matricial con membrana de control de la velocidad de liberación
-
Lámina de soporte exterior
-
Matriz con fentanilo y dipropilenglicol para mejorar el control de la liberación del fármaco
-
Membrana de control de la velocidad de liberación
-
Capa adhesiva de silicona
-
Capa protectora de adhesivo
Buprenorfina
La buprenorfina es un derivado de la tebaína. Su efecto analgésico se debe a su actividad de agonista parcial en los receptores opioides µ[18]. Cuando una molécula se une a uno de estos receptores, éste sólo es activado parcialmente, a diferencia de la morfina que es agonista completo. La alta afinidad de la buprenorfina con los µ-receptores es tal, que los antagonistas opioides para esos receptores, (naloxona) sólo revierten los efectos parcialmente. Por este motivo puede ser utilizada también en el tratamiento de deshabituación de opioides.
Posee una actividad analgésica muy superior (unas 25-50 veces más que la morfina) (0.2 - 0.6 mg intramusculares de buprenorfina, equivalen a 5 - 15 mg intramusculares de morfina). Además su efecto es más prolongado. La depresión respiratoria es dosis-dependiente y equivalente a la de la morfina. La buprenorfina es también un antagonista de los receptores opioides k[19]
En cuanto a la farmacocinética, la buprenorfina se administra por vía intramuscular, intravenosa, sublingual y transdérmica. Se metaboliza por vía hepática siendo el opioide de mejor perfil en el paciente con insuficiencia renal, aunque siempre debemos tener precaución en la rotación de opioides debido a su efecto agonista parcial.
Su absorción es lenta, observándose las concentraciones plasmáticas pico a las dos horas de su administración. Los comprimidos sublinguales de este fármaco ofrecen un efecto analgésico de 6 a 8 horas.
Cuando se usan dosis más elevadas para tratar pacientes con dependencia, la buprenorfina se mantiene efectiva en el organismo entre 24 y 48 horas, llegando a darse casos de hasta 72 horas.
En el tratamiento del dolor crónico es un opioide indicado en pacientes que no toleren la vía oral y aquellos con cierto grado de insuficiencia renal.
Es altamente lipofílica y su biodisponibilidad por vía sublingual es del 50%. Su pico de concentración plasmática aparece tras 3-5 minutos de su administración IM. Su unión a proteínas plasmáticas es alrededor del 96%.
Presenta un elevado volumen de distribución y una vida media de eliminación de 5 horas. La duración de su acción no está relacionada con la vida media de eliminación sino con la vida media de disociación de los receptores. Es excretada inalterada por las heces (68%) y en menor cantidad por la orina en forma conjugada.
La buprenorfina es bien tolerada hemodinámicamente[20], induciendo escasas modificaciones cardiovasculares. Al igual que otros agonistas parciales, la afinidad de la buprenorfina por los receptores opiáceos es superior a la que presentan los antagonistas como la naloxona, por tanto, la depresión respiratoria es más difícil de revertir y generalmente tiene efecto techo tras dosis de 0,3-1 mg. Esto es útil en la clínica para revertir un cuadro de depresión respiratoria provocado por un opiáceo agonista puro sin revertir totalmente la analgesia en los pacientes.
Oxicodona
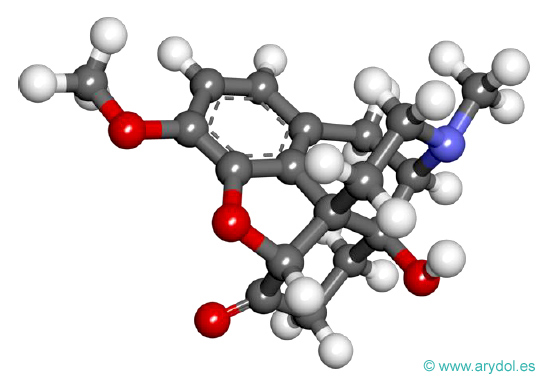
Fig. 17
Fórmula de la oxicodona
La Oxicodona no es, ni mucho menos, un nuevo medicamento. Su síntesis se remonta al año 1916: Martin Freund y Edmund Speyer de la Universidad de Frankfurt (Alemania) sintetizaron varios derivados de la codeína. El compuesto más potente de todos los derivados que se prepararon fue Oxicodona[21].
Oxicodona es un analgésico opioide, con acción agonista pura sobre los receptores opioides del cerebro y de la médula espinal. Recientemente se han descubierto receptores periféricos en intestino, intraarticulares y otros. El efecto terapéutico es principalmente analgésico, ansiolítico y sedante. En cuanto a su farmacocinética, oxicodona posee una biodisponibilidad de hasta el 87% tras administración oral. Tiene un tiempo de semivida de eliminación de 3 horas y se metaboliza principalmente a noroxicodona y oximorfona. La oximorfona posee cierta actividad analgésica pero está presente en el plasma a bajas concentraciones y no se considera que contribuya al efecto farmacológico de oxicodona. La liberación de los comprimidos de liberación controlada (LC) es bifásica[22], con una liberación inicial relativamente rápida, seguida de una liberación más controlada que determina las 12 horas de duración de su acción. El tiempo de semivida de eliminación de oxicodona es 4,5 horas lo que conduce a alcanzar un estado estacionario en aproximadamente 1 día. Durante el tratamiento continuado, el estado de equilibrio (“steady state”) se alcanza al cabo de 24-36 horas horas[23].
Oxicodona es metabolizada por el sistema enzimático citocromo P-450.
Está contraindicada en insuficiencia renal o hepática severa, y se debe usar con precaución en pacientes ancianos. Tiene una potencia doble que la morfina, y se administra en dos tomas diarias.
Disponemos de comprimidos de 5, 10, 20 y 40 mg y en ampollas de 10 mg parenteral.
Oxicodona-Naloxona
Desde hace unos años se dispone en el mercado de una asociación de oxicodona-naloxona que mejora la función intestinal, sobre todo el estreñimiento, sin disminuir su eficacia analgésica. Esto se debe a que la naloxona se absorbe rápidamente fijándose en los receptores intestinales. La dosificación es la misma que sin asociación.
Las características farmacocinéticas de oxicodona en la asociación son equivalentes a las de oxicodona de liberación prolongada. La biodisponibilidad absoluta de oxicodona por vía oral es elevada, hasta un 87%. Una vez absorbida, se distribuye por todo el organismo. Se une a las proteínas del plasma en una proporción aproximada del 45%. Atraviesa la placenta y se puede detectar en leche materna.
La biodisponibilidad sistémica de la naloxona[24] por vía oral es inferior al 3% debido a que se produce un elevado efecto de metabolismo hepático de primer paso. Por este motivo, se supone que el efecto antagonista opiáceo es mínimo y no afecta a la eficacia analgésica de la oxicodona. Previo al paso por el hígado, la naloxona actúa localmente a nivel gastrointestinal bloqueando los receptores opioides y de esta forma reduce el estreñimiento producido por la oxicodona.
Tapentadol
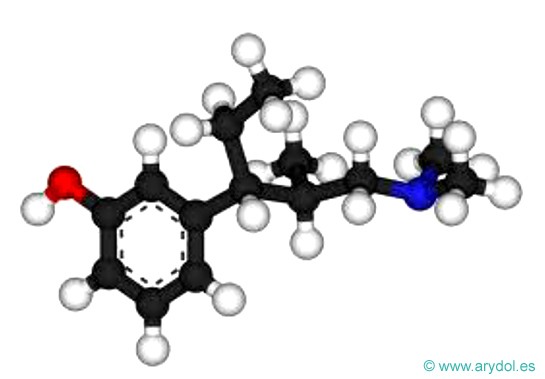
Fig. 18
Fórmula del tapentadol
Este fármaco tiene un mecanismo de acción dual (MOR/NRI), que lo convierte en un opioide especialmente indicado en dolor con componente neuropático y con una menor necesidad de opioide µ (efecto ahorrador µ).
Para entender cómo funciona debemos recordar puntos clave en la fisiología del dolor. Éste se regula mediante vías ascendentes y descendentes; en estas últimas se encuentra el sistema noradrenérgico que siempre es inhibidor y el serotoninérgico con funciones inhibidoras y facilitadoras del estímulo doloroso.
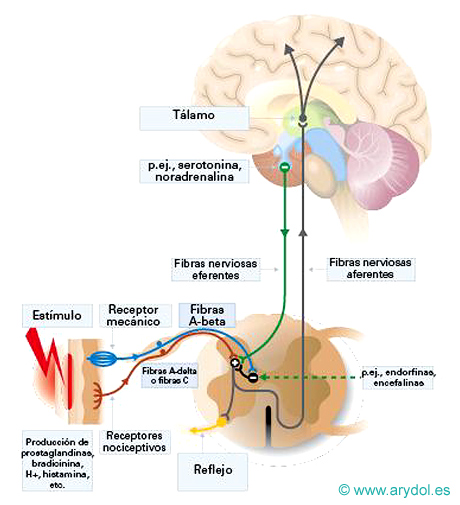
Fig. 19
Uno de los mecanismos de acción del tapentadol es potenciar la acción inhibidora del sistema noradrenérgico descendente (NRI), pero a diferencia del tramadol no es un profármaco, no inhibe la recaptación de serotonian y su metabolismo es simple sin dar lugar a metabolitos activos. Por otro lado tiene una afinidad por el receptor mu opioide (MOR) 50 veces menor que morfina, 5 veces menor que la oxicodona y 20 veces mayor que tramadol. De modo que tiene dos efectos farmacológicos analgésicos complementarios y sinérgicos, tanto a nivel espinal como supraespinal:
- En el asta dorsal de la médula espinal inhibe, tanto a nivel presináptico como postsináptico, la transmisión ascendente del dolor (agonismo µ espinal).
- También se une a receptores µ supraespinales potencia las vías inhibitorias descendentes modificando el balance entre facilitación e inhibición descendente (vía NA / a2-AR)
- De nuevo a nivel medular bloquea la recaptación de NA y potencia la inhibición mediada por a2-AR (inhibición de la recaptación de noradrenalina)
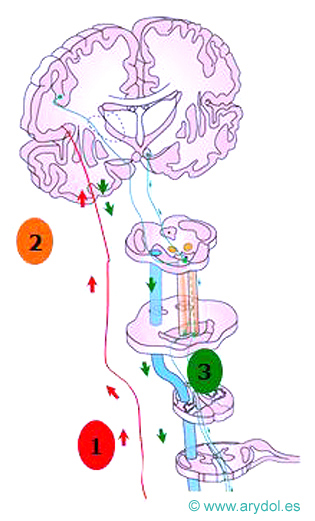
Fig. 20
Acción del tapentadol, a través de las vías ascendentes y descendentes
En dolor agudo predomina el componente opioide y en dolor crónico predomina el componente noradrenérgico. Con el tapentadol, tanto en dolor agudo como en dolor crónico, el sistema opioide y el noradrenérgico se complementan y actúan sinérgicamente; así se caracteriza por su amplia actividad en distintos tipos de dolor, incluido el dolor neuropático y un perfil reducido de efectos secundarios relacionados con receptores mu opioide.
En cuanto a su metabolismo, es hepático de fase 2, mediante O-glucuronidación por múltiples encimas UGT (1A6, 1A9, 2B7) pero no utiliza el citrocromo P450 ni produce metabólicos activos[25,26].
La dosis oral se absorbe rápidamente en un 32% y se distribuye ampliamente. En un 20 % se une a proteínas. Tiene un metabolismo hepático del 97% excretándose luego en la orina en un 99% principalmente en forma de metabolitos conjugados. La vida media es de 4 horas y el efecto máximo se obtiene después de una hora. La duración de acción es 4-6 horas. Sus metabolitos no tienen acción analgésica, de tal modo que la biotransformación del tapentadol por enzimas metabólicas tiene como resultado la inactivación.
Debido a su escasa unión a proteínas plasmáticas tiene un bajo potencial de interacción farmacológica.
Aún es un fármaco relativamente nuevo y hay pocos estudios todavía en comparación con otros fármacos utilizados para el tratamiento del dolor.
Existen varios estudios a destacar; así Schwartz[27] et al. estudian la retirada aleatorizada y controlada con placebo y corroboran la eficacia y seguridad en pacientes con dolor asociado a neuropatía diabética periférica.
Lange[28] et al. realizan un metanálisis para valorar también este fármaco en el dolor crónico artrósico y dolor lumbar en el que concluye que la eficacia de tapentadol es comparable a la de oxicodona, asociándose a un perfil de tolerabilidad gastrointestinal superior y con una incidencia significativamente menor de náuseas, vómitos y estreñimiento cuando se compara con oxicodona.
Los últimos estudios como el realizado por Baron R[29]. et al sobre Tapentadol retard en el dolor lumbar crónico intenso con componente neuropático demuestran que a dosis altas de 500 mg al día la eficacia es similar a la combinación de tapentadol a dosis más bajas de 300 mg asociado a pregabalina 300 mg con una menor incidencia de efectos secundarios sobre el sistema nervioso central.
Hidromorfona
La hidromorfona es una cetona hidrogenada de la morfina sintetizada en Alemania en 1921. La primera referencia clínica sobre la hidromorfona se publica en 1926[30], pero no será hasta 1981 cuando se estudien las propiedades farmacocinéticas y farmacodinámicas de esta molécula[31]. La hidromorfona por tanto es un fármaco bien estudiado como analgésico postoperatorio[32]. Se trata de un opioide que se administra tanto por vía oral como por vía parenteral. Se la denomina con diferentes nombres genéricos, como dihidromorfinona, dihidromorfona, hidromorfinona e hidromorfona.
En cuanto a la farmacocinética, la molécula de hidromorfona es estructuralmente muy similar a la morfina, y difiere de ésta en la presencia de un grupo 6-hidroxilo y la hidrogenación del doble enlace 7-8 de la molécula[32,33]. Se une principalmente a los receptores opioides µ y en menor grado a los receptores δ, sin presentar efectos en los receptores k. La unión a receptores tipo µ es la causa del efecto analgésico, así como de la aparición de efectos secundarios, entre los que se encuentran estreñimiento, náuseas, vómitos, prurito, depresión respiratoria y tolerancia. Asimismo también puede originar retención urinaria, euforia, miosis y dependencia[34,35].
La hidromorfona se metaboliza principalmente en dihidroisomorfina glucurónido e hidromorfona-3-glucurónido (H-3-G). Este último tiene un efecto neuroexcitatorio 2,5 veces más potente que la morfina-3-glucurónido. Se excreta por vía renal en forma de H-3-G en un 35%, sin metabolización en un 6%, como dihidroisomorfina en un 1% y en un 0,1%en forma de dihidromorfina[36].
Su mecanismo “push-pull” permite una única administración diaria; consiste en una cápsula de doble capa osmóticamente activa encerrada en una membrana semipermeable. No tiene metabolitos activos, lo que es una ventaja en pacientes con insuficiencia renal. Su metabolismo no precisa la participación del CYP450, lo que deriva en una menor incidencia de interacciones farmacológicas.
Hay algunos estudios que parecen demostrar una mejora en la calidad del sueño. Está disponible en comprimidos de 4, 8, 16 y 32 mg.
TITULACIÓN EN PACIENTES SIN TRATAMIENTO OPIOIDE PREVIO
Es muy importante la prevención sistemática de náuseas y estreñimiento, en especial en pacientes de riesgo (edad avanzada, sedentarismo); hay que utilizar la vía de administración más adecuada y cómoda para cada paciente. Debe comenzarse siempre por la dosis más baja posible y con los intervalos recomendados para cada opioide:
- MST®: comenzar con 10 mg de Sevredol® cada 4-6 horas, según el dolor. Calcular la dosis total utilizada en 48-72 h. y convertirla en dos dosis diarias de MST®. Ej: Si en 48 h. el paciente ha precisado 80 mg de Sevredol®, pasar a MST®, 40 mg, cada 12 horas y mantener Sevredol® de rescate.
- Oxicodona: comenzar con 5 mg cada 12 horas y aumentar un 50 % cada 48-72 horas hasta alcanzar la dosis óptima, sin efectos adversos graves. Sirve la misma pauta para oxicodona-naloxona.
- Hidromorfona: comenzar con 4 mg cada 24 h y aumentar un 50 % cada 48-72 h. No es un opioide muy adecuado para titulación.
- Tapentadol: comenzar con 50 mg cada 12 horas y aumentar un 50 % cada 48-72 h. hasta obtener alivio del dolor sin efectos adversos notables. (Desde marzo 2013, existe una presentación de 25 mg para ajuste de dosis)
Siempre debe prescribirse un analgésico “de rescate”, de acción rápida o inmediata, para el tratamiento del dolor irruptivo.
ROTACIÓN DE OPIOIDES
En algunos pacientes la tolerancia precoz o ineficacia a dosis razonables nos obliga a plantearnos la rotación de opioide o el cambio en la vía de administración. También es una posibilidad ante la aparición de efectos adversos incontrolados.
Si el dolor predominante es neuropático, se recomiendan, dado su mecanismo de acción, la oxicodona, tapentadol o buprenorfina transdérmica. La última por su efecto antagonista kappa, es una alternativa válida.
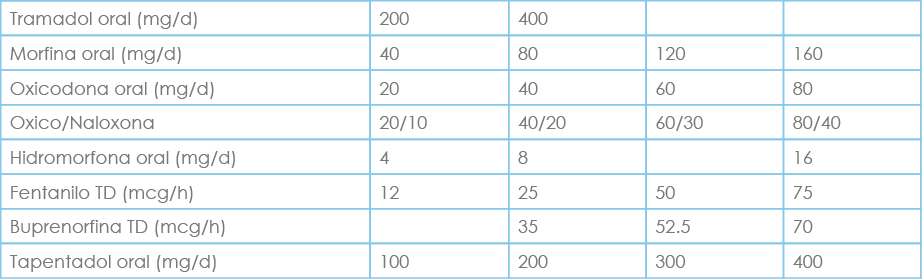
Tabla: Equivalencias de conversión de opioides
En un ejemplo práctico un paciente con morfina de liberación sostenida 200 mg, y de liberación rápida 60 mg (10 mg/4 horas), en total tomaría 260 mg al día, haciendo la conversión a fentanilo TD correspondería aproximadamente a 75 mcg/h; debemos asociar morfina de liberación rápida de rescate (40mg/dosis), revisando posteriormente cuantas dosis ha precisado y ajustando el fentanilo basal.
Siempre debemos titular la dosis y ajustarla de forma individualizada. En los pacientes mayores hay que considerar los cambios farmacocinéticos, la comorbilidad, las posibles interacciones farmacológicas y el fallo orgánico. Si la rotación es secundaria a toxicidad, la dosis debe ser un 30-50% menor[37] que la dosis equivalente del segundo opioide por tolerancia cruzada incompleta, considerando antes de la rotación el tratamiento de los efectos adversos, el descenso de la dosis o el uso de adyuvantes como dexametasona, glicopirrolato u octreótido para el espasmo intestinal[38]. Una vez realizada la rotación puede persistir la toxicidad por un lento aclaramiento del primer opioide. Cuando se deba a dolor no controlado se requieren dosis equianalgésicas; éstas son más variables entre agonistas puros con alta afinidad y agonistas parciales que proporcionarán menor analgesia. La rotación entre los de acción corta y larga tiene que hacerse con cuidado para evitar abstinencia o sobredosis. El 30% de los opioides necesitan una vía alternativa por la presencia de náuseas o mucositis.
En general, el opioide más ampliamente utilizado en las tablas equianalgésicas como referencia es la morfina. Algunos problemas derivados del uso de las mismas son los rangos variables de equivalencia que dan lugar a confusión y a una inadecuada conversión; además esos datos se extraen por computación y no desde ensayos clínicos alejándose bastante del contexto clínico[39,40]. Nunca debemos comparar un opioide puro y una asociación opioide-adyuvante.
GUÍAS EN LA ROTACIÓN DE OPIOIDES
- La rotación secundaria a toxicidad requiere una dosis 30-50 % menor que la dosis equivalente del 2º opioide por tolerancia cruzada incompleta.
- La rotación secundaria a dolor no controlado, requiere dosis equianalgésicas.
- El 30 % de los opioides necesitan una vía alternativa, así como la presencia de náuseas o mucositis.
- Antes de rotar por toxicidad, considerar el tratamiento de los efectos adversos, el descenso de la dosis y la utilización de adyuvantes.
- Considerar los cambios farmacocinéticos en los pacientes mayores, la comorbilidad, las interacciones farmacológicas posibles y el fallo orgánico en la titulación de la dosis.
- Los agonistas parciales proporcionan menor analgesia por incremento de dosis que los agonistas puros con alta afinidad y las dosis equianalgésicas son más variables[41].
- La rotación entre los de acción corta y larga tiene que hacerse con cuidado para evitar abstinencia o sobredosis.
- La rotación en entorno de disfunción orgánica es potencialmente desastrosa, pese a utilizar tablas de equivalencia adecuadas[42].
- Los opioides pueden empeorar el espasmo intestinal. La dexametasona, glycopyrrolato y el octreótido son alternativas válidas.
- La toxicidad inducida por opioides precisa tiempo para su resolución y es posible que la toxicidad persistente tras la rotación ocurra por un lento aclaramiento del primer opioide.
- Rotar a un nuevo opioide antes de alcanzar los parámetros fijados para el primero, es farmacológicamente inaceptable.
SÍNDROME DE NEUROTOXICIDAD INDUCIDO POR OPIOIDES
El síndrome de neurotoxicidad inducido por opioides (NIO) es uno de los efectos adversos del uso de estos fármacos. Se debe a la acumulación de metabolitos tóxicos, como el M3G o el M6G de la morfina. El primero se une a receptores NMDA (activados por el glutamato) por lo que pueden provocar hiperexcitabilidad neuronal, dando lugar a síntomas como alteraciones cognitivas, delirio, convulsiones, mioclonías e hiperalgesia; el M6G, afín a receptores opioides es responsable de los efectos secundarios típicos (somnolencia, depresión respiratoria, alucinaciones, sudoración, coma, miosis, náuseas, retención urinaria). Incluso la propia morfina puede interaccionar con receptores mu y kappa en el hipocampo dando lugar a una toxicidad con síndrome mixto.
Los principales factores de riesgo incluyen dosis altas y/o tratamiento prolongado, alteraciones cognitivas o delirio previo, deshidratación, insuficiencia renal, empleo simultáneo de psicofármacos y pacientes de edad avanzada.
Su manejo incluye principalmente la prevención de su aparición, con el manejo de factores precipitantes; disminución o rotación de opioides y manejo sintomático, intentando mantener siempre un buen control del dolor[43].
¿ADICCIÓN?
El miedo a la farmacodependencia es el hecho que más limita el uso de los opioides.
Recordemos que la dependencia psíquica consiste en el deseo intenso de consumir la droga mientras que la dependencia física da lugar a un síndrome de abstinencia si no utilizamos los opioides para tratar los síntomas. La tolerancia es la necesidad de aumentar la dosis para obtener el mismo efecto. Un paciente adicto tendrá conductas anómalas, una pérdida de control en el uso de la droga, de forma compulsiva, y un consumo continuado a pesar de los riesgos, incluso en periodos asintomáticos.
Existen dos fenómenos derivados del consumo crónico de opioides que obligan a incrementar su dosis, la tolerancia y la hiperalgesia.
En los pacientes con tolerancia se produce un estado de reducción en la potencia de los opioides, que refleja una regularización a la baja o una desensibilización de los pacientes dependientes en el sistema nervioso central (se produce una internalización de los receptores opioides en la membrana celular).
La hiperalgesia inducida por opioides se fundamenta en una elevada respuesta a estímulos nociceptivos tras su administración prolongada debido a la activación de los receptores NMDA que provocan sensibilización espinal.
Para detectar estos problemas debemos monitorizar los pacientes en tratamiento crónico de tercer escalón, siguiendo la analgesia, sus actividades de la vida diaria, cualquier efecto adverso que pueda aparecer o conductas aberrantes actuando en consecuencia, bien modificando la dosis o mediante rotación.
A pesar de las circunstancias descritas, los opioides tienen un papel legítimo con evidencia suficiente para mejorar el dolor y la calidad de vida en pacientes con dolor crónico osteoarticular[44]; el temor a la tolerancia no justifica retrasar su utilización, porque, en ese caso, recurriremos a la rotación o cambio de vía de administración. La adicción es infrecuente (0.5-5%)[45] y si el opioide es el apropiado, la relación beneficio/riesgo siempre es positiva.
TÉCNICAS INTERVENCIONISTAS
Corresponden al cuarto escalón de analgesia de la OMS. Están indicadas en pacientes con mal control analgésico aún con los opioides mayores y las posibilidades de administración ya descritas.
Incluyen infiltraciones de nervios periféricos, de ganglios simpáticos, musculares en cintura escapular o musculatura lumbar profunda, la inyección de toxina botulínica intramuscular e intraarticular, radiofrecuencia convencional (termocoagulación a 60-80º) o pulsada (40º), neuroestimulación y administración intratecal de fármacos.
La neuroestimulación consiste en la aplicación de corriente alterna en las estructuras nerviosas responsables del dolor, evocando parestesia en el área o áreas afectadas. Puede ser medular colocando los electrodos percutáneos en el espacio epidural, en los dermatomas responsables del dolor; está indicado en dolor monorradicular como en síndromes postlaminectomía cervical o lumbar. También se pueden colocar en el territorio de distribución de un nervio periférico (neuralgia occipital, femorocutánea, etc).
El catéter intratecal está dirigido al dolor básicamente oncológico en paciente con expectativa de vida inferior a 3 meses con inadecuado control del dolor por vía oral o parenteral o efectos adversos excesivos. Por esta vía, las dosis de opioides se reducen a la centésima parte de las precisadas por vía oral o parenteral. En pacientes con dolor no oncológico estaría indicado el implante definitivo de bomba intratecal. Se pueden utilizar diferentes fármacos morfina y/o anestésicos locales, baclofeno en el tratamiento de la espasticidad o ziconotida si los opioides son ineficaces.
BIBLIOGRAFÍA
- IASP Pain terms: a current list with definitions and notes on usage. Pain 1986; (Suppl. 3): S215-S221
- Pérez J., Ortiz JR, López S. Anatomía ,fisiología y neurobioquimica del dolor. Anestesia regional y dolor. En Del Olmo C. Aran. 2010; 457-487.
- www.dolopedia.es
- Usunoff KG, Popratiloff A, Schmitt O, Wree A . Functional neuroanatomy of pain. Adv Anat Embryol Cell Biol 2006; 184: 1-115.
- 1 Gracely RH, Lynch SA, Bennett GJ. Painful neuropathy: altered central processing maintained dynamically by peripheral input. Pain 1992; 51(2): 175-94
- Coluzzi PH. The American journal of hospice & palliative care. 1998; 15(1):13–22
- Zaragozá García F. et al.: "Dolor postoperatorio en España. Primer documento de consenso". AEC, GEDOS, SEDAR y SED 2005.
- Coluzzi PH, Schwartzberg L, Conroy JD, Charapata S, Gay M, Busch MA, Chavez J, Ashley J, Lebo D, McCracken M, Portenoy RK. Breakthrough cancer pain: a randomized trial comparing oral transmucosal fentanyl citrate (OTFC) and morphine sulfate immediate release (MSIR).Pain. 2001 Mar; 91(1-2):123-30.
- Portenoy R, Hagen N. Breakthrogh pain: definition, prevalence and characteristics. Pain 1990; 41: 273-81.
- Torres M. Métodos farmacológicos de tratamiento del dolor postoperatorio. Anestesia regional y dolor. En Del Olmo C. Aran. 2010; 457-487.
- Torres LM, Collado F, Almarcha JM et al. Tratamiento del dolor postoperatorio con sistema de PCA intravenoso. Comparación entre morfina, metamizol y buprenorfina. Rev Esp Anestesiol Reanim 1993; 40: 181-84
- Faura CC, Moore RA, Horga JF, Hand CW, McQuay HJ. Morphine and morphine-6-glucuronide plasma concentrations and effect in cancer pain. J Pain Symp Managem 1996; 11: 95-102
- Jacox A, Carr D, Payne R. New clinical practice guidelines for the management of pain in patients with cancer. N Engl J Med. 1993;330:651-5.
- Dalsgaard J, Felsby S, Juelsgaad P, Froekjaer J. Low-dose intra-articular morphine analgesia in day case knee arthroscopy: A randomized double-blinded prospective study. Pain 1994; 56: 151-4
- Wilson JA, Kendall JM, Cornelius P. Intranasal diamorphine for paediatric analgesia: assessement of safety and efficacy. J Accid Emerg Med 1997; 14: 70-2
- Converting to transdermal fentanyl: avoidance of underdosing.Bradley AM, Valgus JM, Bernard S. J Palliat Med. 2013 Apr;16(4):409-11. doi: 10.1089
- Efficacy of Rapid-Onset Oral Fentanyl Formulations vs. Oral Morphine for Cancer-Related Breakthrough Pain: A Meta-Analysis of Comparative Trials. Jandhyala R, Fullarton JR, Bennett MI.J Pain Symptom Manage. 2013 Feb 1. doi:pii: S0885-3924(12)00815-9. 10.1016/j.jpainsymman.2012.09.009.
- M. Carretero Colomer. Dolor crónico intenso. Nuevas perspectivas en el tratamiento. OFFARM. Vol 27, Núm 2, Febrero 2008
- Rodriguez, r, Daza P. Rodriguez MB. Uso de buprenorfina transdérmica en el alivio del dolor por cáncer. Rev. Col. Anest. [online]. Oct./Dec. 2006, vol.34, no.4 [cited 10 October 2008], p.253-257
- Sittl R, Likar R, Poulsen B. Equipotent doses of transdermal fentanyl and transdermal buprenorphine in patients with cancer and noncancer pain: results of a retrospective cohort study. Clin. Ther. 2005; 27 (2): 225-237.
- Sunshine A, et al. Analgesic efficacy of controlled-release oxycodone in postoperative pain. J Clin Pharmacol 1996; 36: 595–603.
- Vondrackova D, Leyendecker P, Meissner W, Hopp M, Szombati I, Hermanns K, et al. Analgesic efficacy andsafety of oxycodone in combination with naloxone as prolonged release tablets in patients with moderate severe chronic pain. J Pain. 2008;9:1144-54.
- Schwartz S et al., Curr Med Res & Opin. 2011; 27(1):151–162
- A randomised controlled trial with prolonged-release oral oxycodone and naloxone to prevent and reverse opioid-induced constipation.Meissner W, Leyendecker P, Mueller-Lissner S, Nadstawek J, Hopp M, Ruckes C, Wirz S, Fleischer W, Reimer K.Eur J Pain. 2009 Jan;13(1):56-64.
- Lange B et al. Adv Ther. 2010; 27(6):381–399
- Obradovic M , Hertel N ,Antoñanzas F. Galvez R. Cost-Effectiveness of Tapentadol in Severe Chronic Pain in Spain: A Cost Analysis of Data From RCTs. Clinical Therapeutics. Volume 34, Issue 4, April 2012, Pages 926–94.
- Schwartz S, Etropolski M, Shapiro DY, Okamoto A, Lange R, Haeussler J, Rauschkolb C. Safety and efficacy of tapentadol ER in patients with painful diabetic peripheral neuropathy: results of a randomized-withdrawal, placebo-controlled trial. Curr Med Res Opin. 2011 Jan;27(1):151-6
- Lange B, Kuperwasser B, Okamoto A, Steup A, Häufel T, Ashworth J, Etropolski M. Efficacy and safety of tapentadol prolonged release for chronic osteoarthritis pain and low back pain. Adv Ther. 2010 Jun;27(6):381-99. doi: 10.1007/s12325-010-0036-3. Epub 2010 Jun 11. Erratum in: Adv Ther. 2010 Dec;27(12):981.
- Baron R., Kern K., Buunen M., Falke D., Steigerwald I. Impact of tapentadol prolonged release versus a combination of tapentadol pr and pregabalin on the neuropathic component of severe , chronic low back pain. Congreso anual de la American Society of Regional Anaesthesia and Pain Medicine. Miami 2012. Ashby MA, Martin P, Jackson KA. Opioid substitution to reduce adverse effects in cancer pain management. Med J Aust. 1999;170:68-71.
- Eddy NB. Dihydromorphinone hydrochloride. JAMA. 1993;100:1032-5.
- Parab PV, Ritschel WA, Coyle DE, Gregg RV, Denson DD. Pharmacokinetics of hydromorphone after intravenous, peroral and rectal administration to human subjects. Biopharm Drug Dispos. 1988;9:187-99.
- Murray A, Hagen N. Hydromorphone "Updates of the clinical pharmacology of opioids". Journal of Pain and Symptom Management. 2005;29:57-66.
- Harvey R, Champe P. Opioid analgesics and antagonists (Lippincott´s illustrated reviews: Pharmacology). Philadelphia: Lippincott; 1992. p. 132-42.
- Ritschel WA, Parab PV, Denson DD, Coyle DE, Gregg RV. Absolute bioavailability of hydromorphone after peroral and rectal administration in humans: saliva/plasmaratio and clinical effects. J Clin Pharmacol. 1987;27:647-53.
- Hang SF, Moore L, Chien YW. Pharmacokinetics and bioavailability of hydromorphone: effect of various routes of administration. Pharm Res. 1988;5:718-21.
- Treatment of cancer pain: Spanish Society of Medical Oncology (SEOM) recommendations for clinical practice. Virizuela JA, Escobar Y, Cassinello J, Borrega P; SEOM (Spanish Society of Clinical Oncology). Clin Transl Oncol. 2012 Jul;14(7):499-504. doi: 10.1007/s12094-012-0831
- Review and critique of opioid rotation practices and associated risks of toxicity. Webster LR, Fine PG. Pain Med. 2012 Apr;13(4):562-70. doi: 10.1111/j.1526-4637.2012.01357.x. Epub 2012 Mar 28. Review. Erratum in: Pain Med. 2012 Dec;13(12):1667.
- Terlinden et al. Eur J Metab Pharmacokinet 32:163 (2007)
- Kneip et al. Drug Metab Letters 2:67 (2008)
- Terlinden et al. Meth Find Exp Clin Pharmacol 32:31-38 (2010)
- Nagar y Raffa, J Fam Pract 57 (Suppl 6A): S1-S8 (2008)
- Shanee PA, Walsh D, Lasheen W, Davis MP, Lagman RL. Opioid equianalgesic tables: are they all equally dangerous? J Pain Symptom Manage. 2009 Sep;38(3):409-17.
- Cid , M L. Síndrome de neurotoxicidad inducido por opioides (NIO). Rev Soc Esp Dolor 15 (2008)
- Sánchez, Espinosa y Marcos. Actualización en el dolor crónico osteoarticular con opioides en AP. Monográficos SEMG. Enero 2012
- Bob Kwok Bun Chan et als. Expert Opin. Opioids in chronic non-cancer pain. Pharmacother 2011;12(5):705-720